






























In medical device development, two critical stages of the design process are often confused: design verification and design validation. What are their distinct roles in the delivery of quality products, and why are the processes so prone to confusion and collapse?
Design verification and design validation: what’s the difference?
Here’s the classic definition:
- Design verification ensures you’ve built the product right.
- Design validation ensures you’ve built the right product.

Why is design V&V so important?
Medical device companies and customers have a lot at stake if the V&V process isn’t right.
It’s worth remembering that nearly 10% of all FDA warning letters in the last five years have resulted from inadequately defined and executed validation processes.
Without a systematic process of checks against a formal list of engineering specifications and user requirements, developers can never be certain that their finished product:
- Will function correctly according to requirements.
- Will do what is intended across its required lifespan.
That’s why V&V sits at the core of most medical device quality standards and regulations.
V&V in Medical Device Regulation
Validation and verification are formal parts of the design control requirements for the world’s major medical device development regulations and standards.
- FDA 21 CFR Part 820 (Quality System Regulation) in the USA.
- ISO 13485:2016 - International QMS standards.
- ISO 14971 - International risk management standards.
- European Union Medical Device Regulation (EU MDR).
- Europen Union In Vitro Diagnostic Device Regulation (EU IVDR).
- IEC 62304 - Quality standard specific to medical device software.
It’s safe to say you cannot successfully launch a medical device for sale around the world without making provision for V&V.
What does the FDA V&V process require from developers?
The FDA helpfully supply a diagram illustrating exactly how the design verification and validation process fits in a typical sequence of design controls.
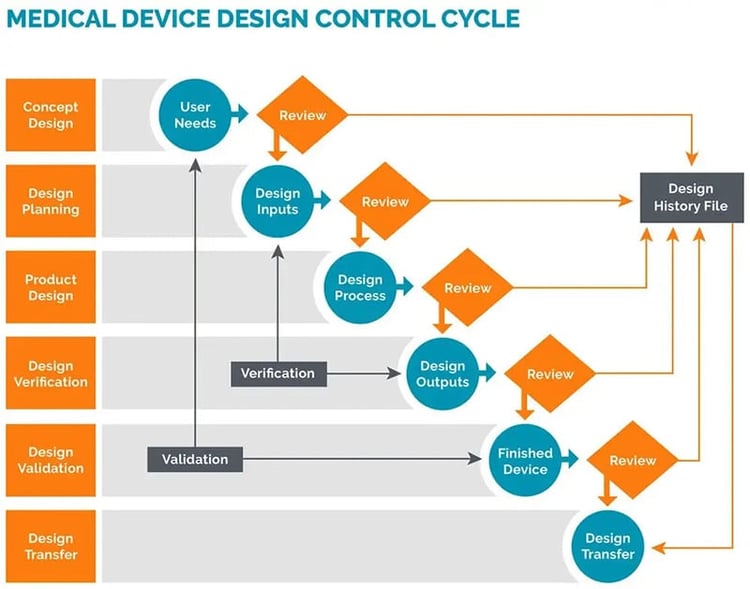
What is design verification?
According to the FDA, design verification is “confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.”
How does that work in practice?
In the classic design and development model, user requirements are gathered at the start of a process. These are then converted into documented design inputs, which then can be turned into design outputs.
Design inputs may include:
- Functional requirements.
- Safety considerations.
- Regulatory requirements.
- Risk control requirements.
Design outputs may include:
- Engineering specifications.
- Bills of material.
- Engineering schematics.
- Device blueprints.
How do design inputs become verified design outputs?
The FDA say that developers must have a systematic process for documenting design inputs and generating appropriate design outputs, with a means of testing that their solution can meet these requirements.
This requires formal planning for design verification as the development process continues.
Businesses should have the tools to create formal processes and workflows that allow developers to collaborate on Functional Requirement documentation, systematically approve design outputs against specifications, generate evidence of verification - and iterate and improve as required.
These workflows should allow for planning and evidence of verification of each design and development phase, with STOP/GO moments to assess whether outputs match required deliverables before the next phase is triggered.
In this way, evidence of how inputs are turned into outputs -then verified and approved by key stakeholders - can be captured and stored as evidence for the Design History File.
Traceability matrices
The FDA like developers to use traceability matrices as a way of visualising and centralising their verification process, ensuring they can successfully map user needs to functional requirements and engineering requirements.
As an example, here’s how one user requirement for a mobile hospital ventilator might become a tangible (and verifiable) design input and output:
Design Input
The ventilator shall run on battery power at maximum settings for a minimum of 90 minutes.
Design output
The ventilator will run on a lithium-ion battery pack rated for at least 100 Amp Hours.
This, in turn, can create a number of test cases to establish that the design requirement has been met. For this example, a suitable test case may include:
Test case
Verify run-time on maximum settings with a fully charged new battery.
Documenting all this within a matrix allows developers to systematically create a traceable connection between key requirements and key design decisions, plus link to evidence that the design solution can function as required. All in a way that will be auditable in the future.
But the design verification process only concentrates on checking that the product is being designed and built as required. It does not validate the product as a whole.
What are the key differences between the design verification and validation process
|
Verification |
Validation |
|
Scope: Design Outputs meet Design Inputs |
Scope: The design meets user needs and the intended use of the device |
|
System, subsystem and unit testing. |
Test focused at a system level to demonstrate usability, safety and efficacy in the field |
|
During development |
After Development |
|
Can be performed on prototype devices |
Must be performed on the same devices as the ones that will go on the market or equivalent devices |
|
Performed against specific requirements selected by the owner of the design |
Performed against the selected user needs and requirements established by international standards. |
Why a design verification process alone is not enough to ensure quality
Design verification is about turning functional requirements into functioning design specifications. It does not assess the finished product as a whole against user needs or look at its performance when it’s in customers’ hands. And that presents a problem.
After all, it’s possible for a product to be designed precisely to meet identified functional requirements but fail usability tests in the real world. For example, when the product is handled in the field, it might prove unusable in one significant way by certain patients, or it may malfunction after continued deployment.
When the verification process is not enough
The Medtronic SynchroMed Infusion Pump had to be recalled due to motor corrosion and battery issues following use by actual patients. This highlights the crucial importance of extensive reliability testing and assessment of long-term performance in real-world conditions as part of a validation process.
What is design validation?
So, to catch these kinds of potential quality issues, the finished product itself must be validated against a comprehensive list of user requirements.
For the FDA, validation means “confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled.” In other words, will it work in the real world as expected and required by the people who will use it?
To ensure this, specific tests and processes must be devised that capture the product's performance under varied conditions, ensuring it meets all necessary usability, safety, efficacy, and quality tests. This comprehensive approach is crucial for guaranteeing the reliability and effectiveness of the product in real-world applications.
Tests, analysis and process that might be used include:
- Usability testing.
- Clinical Investigation.
- Biocompatibility Evaluation.
- Electrical Safety and Electromagnetic Compatibility, in case of active device.
- Packaging Validation / Sterilisation Validation.
Where can it all go wrong?
For modern medical devices, the sheer weight of documentation generated as part of the requirements gathering, design iteration and verification phase can be overwhelming.
Developers can easily lose track of their design decision-making in a tsunami of different documents. Without the right software, as they build their Design History File to share with auditors, they might be unable to find previous iterations of documents. The locations of test results in their traceability matrices may become broken or missing. This can be a serious problem if you need to show exactly what V&V activity was conducted, when and by whom.
4 tips for more efficient design verification and validation
1. Start planning V&V process early
As you gather your user needs and functional requirements, consider how they will be verified and validated so you can properly plan required activity and leave enough time to do so.
Establish your verification and validation matrices at the start of the process and ensure you have the tools to manage change control within them.
2. Choose the right digital tools
Make sure you have the right digital tools to create workflows that will generate required V&V evidence. The right eQMS (electronic quality management system) will help you gather and collate user requirements and other design inputs that can be shared with developers to produce required design outputs. Approval mechanisms should automatically trigger sequences of V&V activity collating evidence of compliance for inclusion in your DHF.
3. Ensure V&V matrices are controlled
You need the digital tools to ensure that V&V matrices cannot be changed without authorisation and that they are always pointing to the right place. With the right eQMS, you should be able to set up links to plans, test results or usability studies that cannot be broken. As you iterate and update this documentation, your matrices should link to the latest version of each - with audit histories available for each that show their evolution.
4. Remember Verification and validation is an ongoing process
The design verification and validation process is always ongoing. As post-surveillance activity continues and you consider new releases of your product, you may need to change aspects of the design. When you do so, the V&V processes need to be retriggered to test and provide evidence for the efficacy and safety of proposed and implemented changes. An eQMS will help you accelerate and automate this process as you work on your next product iteration.
Modern medical devices are complex and multifunctional. The requirements for verification and validation can produce sprawling documentation that can easily become unmanageable. To meet these demands, you can’t rely on DIY approaches with shared Google Drives and Excel documents. You need the tools to manage and automate design workflows. You need the digital tools to help you plan your processes while organising and collating documentation for approval by auditors.



%20(1).webp?width=133&height=76&name=ISO%20IEC%2027001%20(1)%20(1).webp)