





























 A QMS should ultimately help guard against customer harm or dissatisfaction, by guarding against the risk of failure of the design and build processes. In turn, this helps avoid the commercial failure of the product itself, too. But how do you achieve this? In this article, you’ll read about medical device risk management and how a QMS can support a risk-based approach. We’ll also share 5 benefits of adopting a risk-based approach to quality management in ISO 13485:2016.
A QMS should ultimately help guard against customer harm or dissatisfaction, by guarding against the risk of failure of the design and build processes. In turn, this helps avoid the commercial failure of the product itself, too. But how do you achieve this? In this article, you’ll read about medical device risk management and how a QMS can support a risk-based approach. We’ll also share 5 benefits of adopting a risk-based approach to quality management in ISO 13485:2016.
What Is Medical Device Risk Management in ISO 13485?
The purpose of medical device risk management practices are to minimise the risk either to the achievement of the objectives of the med tech project or to the reliability of the developed device and safety of the users.
In any medical device development project a Risk Management Plan needs to be prepared. This plan lays out the approach to identifying, tracking and resolving each risk to a device, associated with its design, production, storage and usage.
The identification of hazards will also need to be documented in the Hazard Identification Document (HID).
All of these documents will ultimately be contained within the “Risk Management File”. And the elements of the plan should be implemented and evident according to the procedures of the QMS.
What Is the Risk-Based Approach in ISO 13485?
Risk is a hot topic in ISO 13485: 2016. It’s mentioned much more frequently in the current documentation than in the standard’s previous iteration (10 times more often, to be exact).
ISO 13485 design control describes a developers’ responsibility in this area as an overarching risk prevention strategy. But it also acknowledges that this ‘risk-based thinking’ should be commensurate with the level of threat of harm posed by non-conformance in each area:
“The organisation shall apply a risk-based approach to the control of the appropriate processes needed for the Quality Management System”
Elsewhere the approach to risk is defined as:
“The systematic application of management policies, procedures and practices to the tasks of analysing, evaluating, controlling and monitoring risk”
Helpfully, the regulation draws our attention to areas of particular importance here, including design and development, training and purchasing (i.e. working with third party suppliers).
These are areas where there is particular danger posed by non-conformance (e.g. a malfunction in design) or where there is increased risk of losing control over a particular area of compliance, for example in training or outsourcing.
The responsibility for assessing the compliance of your suppliers against the relevant regulations might seem an onerous one, but it is entirely consistent with the risk-based approach. And you will need to have a process in place to assess whether their QMS (and, therefore, any product or service they are supplying to you) is compliant.
But this risk-based approach, with its focus on outcomes and reducing risk of customer harm or dissatisfaction, can also be an opportunity.
It’s intended, after all, to move away from an ‘inspect and control’ regulatory paradigm for the improvement of quality in the medical device industry and save companies the effort of endless remedial work.
It should mean you are continually reviewing processes and focused on improving the consistency and quality of your end product before anything goes wrong. This is an opportunity for innovation as you strive to make your processes as efficient, innovative and error free as possible:
5 Key Benefits of Risk-Based Thinking in ISO 13485
- Improved levels of regulatory compliance
- Process efficiency improves when attention is placed on higher risk issues and requirements and dialed down for lower risk items
- Enables companies to focus efforts on the aspects of the QMS with the highest risk
- Ensures problems are solved before they impact the product – reduces costs of remedial work
- Innovation through the effort of continual improvement
How Risk Management Is Built into Medical Device Design
One of the harmonised standards associated with ISO 13485 is ISO 14971 – “Application of Risk Management to Medical Devices”.
Those requirements are clearly stipulated within ISO 13485 itself.
Clause 7.1 of ISO 13485: 2016 specifies the way the risk management requirements of the QMS should be implemented as you begin to formally design and build.
It describes a mandatory process for planning and product realisation, which governs, captures and records every material stage of the design and development of a medical device. These processes are put in place to minimise the risk of failure, while promoting transparency and accountability at all times. And you need to plan for this.
ISO 13485 expects you to create a medical device design process that:
- Breaks the process down into stages and with identifiable deliverables at each stage
- Identifies check points for reviews and specifies its participants
- Establishes a communication plan and a mechanism for communications
- Creates and updates necessary records as the process continues
As part of the way risk is managed throughout the process the QMS must also make provision for proper design change control; devising processes that assess the potential risks involved in making alterations. To this end ISO requires that you:
- Document the design changes
- Approve the changes only after review and verification
- Evaluate the effects of changes
- Document the results, and take any necessary corrective and preventive actions to solve problems arising from them
And as the diagram below shows, almost the entire product development lifecycle of a medical device needs to be governed and controlled by the kind of QMS it specifies:
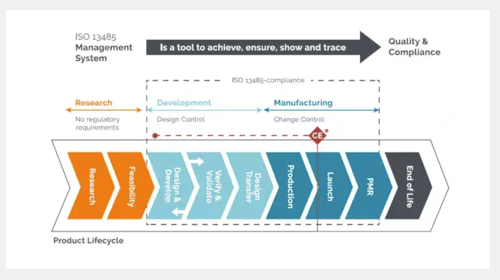
Proper medical device risk management will take time and effort. It will take considerable time and effort to create and define the SOPs that form your QMS as a whole, as well as building the system of digital or real world files where all its outputs will reside.
It will be expensive, time-consuming, and even impossible to piece all that together retrospectively.
But the reality is you will not be able to deliver or legally market your product without doing this.
Want to Build a Medical Device? Start Here
If you want to be a medical device developer you need a great idea for a product, as well as the ability to build it. But if you want to be a successful medical device developer, you're going to need more. In the guide below, you’ll find everything you need to design and build a medical device, and how to bring it to market using the right QMS.



%20(1).webp?width=133&height=76&name=ISO%20IEC%2027001%20(1)%20(1).webp)