





























 When developing a healthcare product, it's crucial to determine its classification: Medical Device (MD), In Vitro Diagnostic Device (IVD), Active Implantable Medical Device (AIMD), or Software as a Medical Device (SaMD). This classification impacts the regulatory approval process necessary for market entry.
When developing a healthcare product, it's crucial to determine its classification: Medical Device (MD), In Vitro Diagnostic Device (IVD), Active Implantable Medical Device (AIMD), or Software as a Medical Device (SaMD). This classification impacts the regulatory approval process necessary for market entry.
What’s the definition of a medical device?
According to the U.S. Food and Drug Administration (FDA), a medical device encompasses:
"Any instrument, machine, contrivance, implant, in vitro reagent that's intended to treat, cure, prevent, mitigate, or diagnose disease in humans."
This broad definition includes both physical instruments and standalone software, highlighting the diverse nature of medical devices.
“any instrument, machine, contrivance, implant, in vitro reagent that's intended to treat, cure, prevent, mitigate or diagnose disease in man”.
Leaving aside the historic sexism of the wording this is a definition that is echoed by regulatory bodies around the world.
It reminds developers that as far as regulators are concerned it’s not just hardware that is capable of being a medical device, but instead ‘any contrivance’ - which can include stand-alone software as much as physical instruments.

How does the EU MDR define a medical device?
The European Medical Devices Regulation, uses a similar definition of a medical device to the FDA and goes on to define the various intended purposes of medical devices as:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease;
- diagnosis, monitoring, treatment, alleviation of, or compensation for an injury or disability;
- the investigation, replacement or modification of the anatomy or a physiological or pathological process or state;
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations;
- for the control or support of conception; or
- products specifically intended for the cleaning, disinfection or sterilisation of devices.
The term ‘medical device’, then, covers the full spectrum of machines and contrivances from IVD and implantables to standalone software.
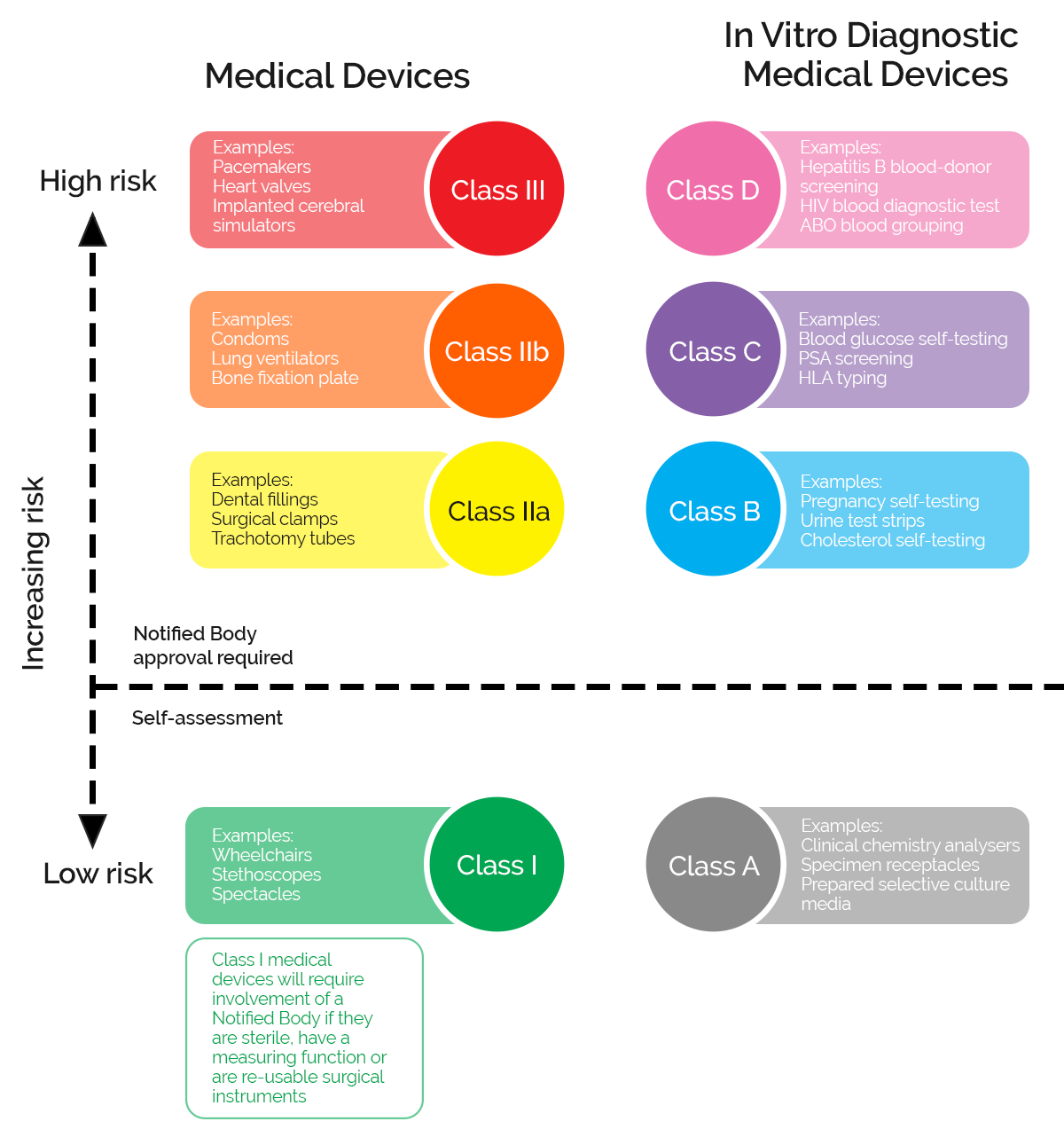
But these different types of medical devices can pose different levels of risk to end users, so most regulatory regimes break them down into further distinct categories and classifications.
In the EU these risk-based classifications then determine the kind of quality management and approval requirements you need to go through to launch your product onto the market.
What is an In Vitro Diagnostic medical device?
From the Latin meaning “within the glass”, in vitro diagnostic devices refer to tests used on biological samples outside the body, to determine state of health. The EU IVDR is the legislation that regulates the production of these devices, it defines IVDs (In Vitro Diagnostic Devices) as:
'Any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body."
IVDs came to prominence in the worldwide Covid pandemic where millions of self-testing kits were procured and distributed by governments around the world. There is now great interest in self-testing technology as a game-changing moment for public health to increase early disease detection and improve the monitoring of chronic conditions.
It’s also presenting new opportunities for challenger tech companies who are bringing novel software to the market and developing wearables to enhance and improve sample gathering and diagnostic capabilities. These companies need to pay particular attention to how their devices will be classified and regulated so they can build QMS (Quality Management Systems) that are equal to the task.
What is an Active Implantable Medical Device (AIMD)?
The EU define an AIMD as:
“a device which, for its functioning, relies on electrical energy or a power source other than that directly generated by the human body or gravity) intended to be totally or partially introduced, surgically or medically, into the human body”
Examples of AIMD include pacemakers, defibrillators, cochlear implants and neurostimulators. This category offers some of the most interesting opportunities for IoT specialists from different sectors to bring their insights and miniaturised technology to help treat some of the most serious and complex medical conditions. Again, challenger tech firms need to be prepared for the demanding quality management requirements entailed in developing high risk medical products
What are the classifications of medical devices in the EU?
In the EU medical devices and IVDs are broken down into different classifications reflecting the level of risk they pose to their users.
In the EU, manufacturers need to demonstrate that the medical device meets the requirements of the MDR or IVDR by carrying out Conformity Assessments (CA). The assessment route depends on the classification of the device and may require your Quality Management System to be audited by one of the Notified Bodies which are licensed and approved by regulators in each country.
The lowest risk devices can be self-certified with notified bodies, but the greater the risk profile the more extensive the QMS and auditing requirements become. For Class 3 devices, using novel or cutting-edge technology, these may include evidencing exhaustive clinical trial and hosting facilities inspections that can stretch your development and approval timeline out by many years.
In the US a different regime maintains which you can read about in this blog covering the FDA classification and submission process:

But where does all this leave software as a medical device?
When is software classed as a medical device (SaMD)?
Software as a Medical Device (SaMD) is defined as:
“Software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device."
In the EU regulation SaMD is referred to as Medical Device Software (MDSW)
Is an app a SaMD?
Apps or devices that monitor fitness, health or general wellbeing, are not usually considered medical devices. Nor are those that simply digitise or organise information that would normally be printed or completed by hand (such as patient diaries for recording blood pressure).
At the other end of the scale is software that is part of or embedded into a medical device. This is not regarded as SaMD because its safety and efficacy will be demonstrated and evidenced to regulators as part of an overall device submission.
However, many other novel apps and pieces of stand-alone software are classified as medical devices and will be subject to compliance requirements as discussed above.
 Examples of SaMD
Examples of SaMD
Standalone software becomes a medical device when it starts to inform or direct diagnoses and treatment for a range of conditions. If you are unsure how the regulators will classify your particular product you should talk to a consultant, a Notifying Body or the regulator direct.
Examples of SaMD include:
- Software intended to measure and transmit blood glucose levels, calculate insulin dose required and drive an insulin pump to administer the calculated dosage
- Software that allows clinicians to view images from an MRI machine on a smartphone as part of their diagnostic process
- Software that performs image post-processing and analysis to help detect tumors or breast cancer
- Software that collects data from multiple sources (x-rays, scans, etc.) and turns that data into 3D models.
- Software that collects data, then uses algorithms to develop a treatment plan for a specific condition.
- Software intended to track a user’s health information, including blood pressure. The software analyses the information in order to provide a diagnosis of hypertension and risk of cardiac disease.
Just as with other medical devices, as an SaMD’s risk profile increases so does the intensity of regulatory scrutiny. For example, those gathering and analysing critical data for the treatment of chronic conditions will be subject to the most rigorous evidencing and approval processes.
| State of patient condition | Significance of information provided by the MDSW to diagnosis/therapy | ||
| High | Medium | Low | |
| Critical condition | Class III | Class IIb | Class IIa |
| Serious condition | Class IIb | Class IIa | Class IIa |
| Non serious condition | Class IIa | Class IIa | Class IIa |
But what about Brexit?
Up until recently, the United Kingdom used the same legislation and terminology as the EU. However, due to Brexit, new rules apply in Great Britain and Northern Ireland. Manufacturers wishing to place medical devices on the Great Britain market will need to follow the new UKCA rules to identify and classify their devices (including SaMD).
But existing CE marks will continue to be recognised in Great Britain until 1st July 2024. CE marking will continue to be required in Northern Ireland where the EU MDR and IVDR will still apply.
Conclusion
Understanding your medical device type and classification is critical to figuring out the kind of clinical investigations and quality management systems you will need in place to launch successfully in your target market. It will dictate the intensity of regulatory scrutiny and the length of time it will take you to get to launch. As more novel devices enter the market and challenger companies shift into the space, ensuring you understand the detail of the regulation and how it applies to you may save you from having to rework schedules, find extra funding, and backsolve complex compliance requirements in the future.



%20(1).webp?width=133&height=76&name=ISO%20IEC%2027001%20(1)%20(1).webp)